Mitochondriale Myopathien
Was sind Mitochondrien?
Die Mitochondrien sind Organellen (Strukturen der Körperzellen mit bestimmter Funktion), die für die Energiegewinnung der Zellen verantwortlich sind. Man kann sie als „Kraftwerke“ der Zellen bezeichnen. Sie stellen fast 90% unseres gesamten Energiebedarfs bereit.
In der sog. Zellatmung findet die „Verbrennung“ von Sauerstoff statt. Dazu gibt es die sog. „Atmungskette“ an der inneren Membran der Mitochondrien. Darüber hinaus haben die Mitochondrien aber noch weitere Funktionen, so sind sie z.B. auch für den Abbau der Fettsäuren verantwortlich.
Mitochondriopathien als vielgestaltige Erkrankungen
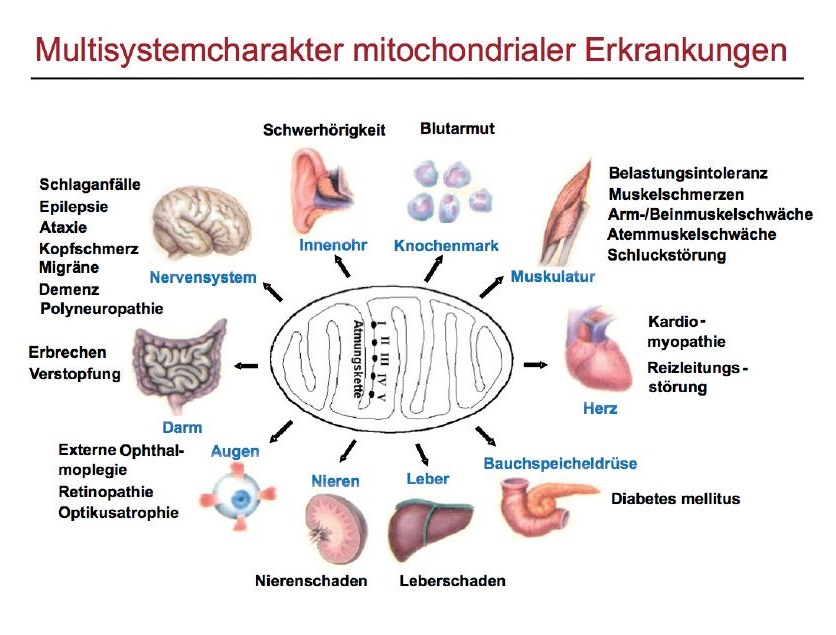
Mitochondriale Erkrankungen sind Erkrankungen, bei denen ein Defekt in den Mitochondrien vorliegt, der in der Regel eine genetische Ursache hat. Funktionsstörungen der Mitochondrien betreffen insbesondere die Muskelzellen, da diese einen hohen Energiebedarf aufweisen. Es kann zu einer Mitochondrialen Myopathie (Muskelerkrankung) kommen.
Aber auch andere Zellen und Gewebe können betroffen sein, so z.B. das Nervensystem, das Auge und das Innenohr. Störungen des Magen-Darm-Traktes, der Leber oder der Bauchspeicheldrüse u. a. kommen ebenfalls vor. Bei Erkrankungen der Mitochondrien handelt es sich daher oft um sog. Multisystemerkrankungen, bei denen verschiedene Organe erkranken.
Vorkommen und Symptome
Eine Mitochondrien-Erkrankung kann sowohl im Kindesalter als auch im Erwachsenenalter auftreten, die klinische Ausprägung kann im Verlauf wechseln.
Häufig berichten Patienten mit Mitochondrialen Myopathien über eine belastungsabhängige Muskelschwäche, chronische Schmerzen, allgemeine Erschöpfung und Belastungsintoleranz. Aber auch eine andauernde Muskelschwäche ist möglich.
Wegweisend für eine Mitochondriale Myopathie ist, wenn die Augenmuskeln betroffen sind. Bei manchen Patienten kann eine Herzmuskelschwäche auftreten, aber auch Störungen der Nervenleitung im Herz sind möglich, was zu Herzrhythmusstörungen führen kann. Ist das Gehirn betroffen, können z.B. epileptische Anfälle, geistige Behinderungen oder schlaganfallähnliche Beschwerden auftreten. Die Nerven von Armen und Beinen können ebenfalls betroffen sein. Am Auge können besonders der Sehnerv und die Netzhaut erkranken. Außerdem findet sich häufig eine Schwerhörigkeit und eine Zuckerkrankheit.
Typische Kombinationen von Symptomen können den Arzt zur Diagnose der Mitochondrialen Erkrankung führen. Man spricht dann von charakteristischen Syndromen. Allerdings weisen vielfach Patienten nicht das volle Bild dieser Syndrome auf und es gibt manchmal auch eine Überlappung zweier Syndrome. Mitochondriale Erkrankungen gehören zu den seltenen Erkrankungen, je nach Syndrom auch zu den sehr seltenen Erkrankungen.
Weiterführende Infos
Im Download finden Sie Informationen zur genetischen Grundlage, zur Diagnosestellung und zu Therapiemöglichkeiten. Auch eine Informationsbroschüre für Berufsgruppen, die mit mitochondrial erkrankten Kindern arbeiten sowie einen umfangreichen Patientenratgeber können Sie hier herunterladen.
Forschung zu Mitochondrialen Myopathien
Infos und Selbsthilfeangebote
Unsere Diagnosegruppe vertritt die Interessen der Betroffenen und Angehörigen.
Mito-Gruppe Mitochondriale Erkrankungen in der DGM