Amyotrophe Lateralsklerose (ALS)
Amyotrophe Lateralsklerose (ALS)
Die Amyotrophe Lateralsklerose ist eine sehr ernste Erkrankung des zentralen und peripheren Nervensystems. Sie ist seit mehr als 100 Jahren bekannt und kommt weltweit vor. Ihre Ursache ist mit Ausnahme der seltenen erblichen Formen bisher unbekannt. Die Abkürzung für Amyotrophe Lateralsklerose ist ALS. ALS hat nichts mit MS (Multiple Sklerose) zu tun, es handelt sich um zwei völlig unterschiedliche Erkrankungen.
Pro Jahr erkranken etwa ein bis zwei von 100.000 Personen an ALS. Die Krankheit beginnt meistens zwischen dem 50. und 70. Lebensjahr, nur selten sind jüngere Erwachsene betroffen. Männer erkranken etwas öfter als Frauen (1,6:1). Die Häufigkeit der ALS scheint weltweit etwas zuzunehmen. Das Krankheitstempo ist bei den einzelnen Patienten sehr unterschiedlich, die Lebenserwartung ist verkürzt.
Die ALS betrifft nahezu ausschließlich das motorische Nervensystem. Die Empfindung für Berührung, Schmerz und Temperatur, das Sehen, Hören, Riechen und Schmecken, die Funktionen von Blase und Darm bleiben in den meisten Fällen normal. Leichtgradige, meist nur in speziellen Tests feststellbare Einschränkungen der geistigen Leistungsfähigkeit können auftreten, etwa 5 % der Patienten entwickeln das Bild einer frontotemporalen Demenz mit Auffälligkeiten im Bereich der Kognition und des Verhaltens.
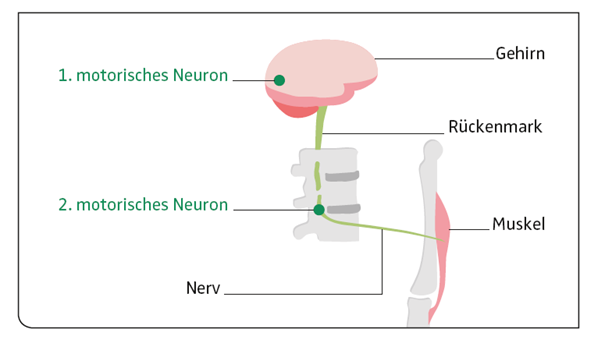
Das motorische System, das unsere Muskeln kontrolliert und die Bewegungen steuert, erkrankt sowohl in seinen zentralen („oberes oder 1. Motorisches Neuron“ im Gehirn mit Pyramidenbahn bis ins Rückenmark) als auch in seinen peripheren Anteilen („unteres oder 2. motorisches Neuron“ in Hirnstamm und Rückenmark mit den motorischen Nervenfasern bis zum Muskel).
Die Erkrankung der motorischen Nervenzellen im Rückenmark und ihren Fortsätzen zur Muskulatur führt zu unwillkürlichen Muskelzuckungen (Faszikulationen), Muskelschwund (Atrophie) und zu Muskelschwäche (Paresen) an Armen und Beinen und auch in der Atemmuskulatur. Wenn die im Hirnstamm liegenden motorischen Nervenzellen betroffen sind, ist die Sprech-, Kau- und Schluckmuskulatur geschwächt.
Die Erkrankung der motorischen Nervenzellen in der Hirnrinde und ihrer Verbindungen zum Rückenmark führt sowohl zu einer Muskellähmung wie zu einer Erhöhung des Muskeltonus (Spastik) mit einer Steigerung der Reflexe.
Welche Symptome zeigt die ALS?
Die ersten Symptome können bei den einzelnen Kranken an unterschiedlichen Stellen auftreten. Muskelschwund und -schwäche können sich z.B. zunächst nur in der Hand- und Unterarmmuskulatur einer Körperseite zeigen, bevor sie sich auf die Gegenseite und auf die Beine ausdehnen. Seltener ist ein Beginn in der Unterschenkel- und Fußmuskulatur oder in der Oberarm- und Schultermuskulatur. Bei einem Teil der Erkrankten treten erste Symptome im Bereich der Sprech-, Kau- und Schluckmuskulatur auf (Bulbärparalyse). Sehr selten äußern sich die ersten Symptome in Form von spastischen Lähmungen. Schon in den Frühstadien der ALS wird häufig über unwillkürliche Muskelzuckungen (Faszikulationen) und schmerzhafte Muskelkrämpfe geklagt.
In der Regel schreitet die Krankheit über Jahre gleichmäßig langsam fort, dehnt sich auf weitere Körperregionen aus und führt zu einer zunehmenden Atemschwäche. Die Lebenserwartung ist im Mittel auf drei bis vier Jahre verkürzt. Sehr langsame Verläufe über 10 Jahre und mehr sind jedoch bekannt.
Wie wird die ALS diagnostiziert?
Zuständig für die Diagnosestellung ist der Neurologe. Der Patient wird zunächst klinisch untersucht, insbesondere muss die Muskulatur im Hinblick auf Muskelschwund und Kraft sowie Faszikulationen beurteilt werden. Ebenso ist eine Beurteilung von Sprache, Schluckakt und Atemfunktion wichtig. Die Reflexe müssen geprüft werden. Darüber hinaus müssen andere Funktionen des Nervensystems, die von der ALS üblicherweise nicht betroffen sind, untersucht werden, um ähnliche, aber ursächlich unterschiedliche Erkrankungen zu erkennen (sog. ALS-Mimics) und um Fehldiagnosen zu vermeiden. Der Ausschluss anderer Erkrankungen ist wegen der meist besseren Prognose und Behandelbarkeit unbedingt notwendig.
Eine wichtige Zusatzuntersuchung ist die Elektromyographie (EMG), die den Befall des unteren Motoneurons auch in klinisch noch normal erscheinenden Muskeln beweisen kann. Untersuchungen der motorischen und sensiblen Nervenleitung dienen der Abgrenzung zu Erkrankungen der peripheren Nerven (Neuropathien), z.B. der klinisch zu Beginn täuschend ähnlichen multifokalen motorischen Neuropathie (MMN).
Zusätzlich zum EMG wird zunehmend der neuromuskuläre Ultraschall eingesetzt, mit dem schmerzfrei Faszikulationen und Muskelatrophien erfasst werden können.
Außerdem sind umfassende Untersuchungen des Blutes und des Urins notwendig, insbesondere um immunologische oder andere entzündliche Erkrankungen, Stoffwechselstörungen und hormonelle Störungen (z.B. Schilddrüse) auszuschließen. Bei der Erstdiagnostik ist auch eine Untersuchung des Nervenwassers (Liquor) sinnvoll. Derzeit wird intensiv nach sogenannten Biomarkern gesucht, mit deren Hilfe früh im Blut oder Nervenwasser die Diagnose gestützt werden kann. Bereits frühzeitig sind bei der ALS erhöhte Neurofilament-Konzentrationen in Liquor und Serum nachweisbar, dies ist jedoch nicht spezifisch für die ALS, sondern kommt auch bei anderen neurologischen Erkrankungen vor. Eine Muskelgewebe-Untersuchung ist im Regelfall nicht erforderlich und wird nur für diagnostisch besonders schwierige Fälle empfohlen zur Abgrenzung anderer neuromuskulärer Erkrankungen.
Bildgebende Untersuchungen gehören ebenfalls zur Diagnostik. Eine Kernspintomographie (MRT) des Kopfes und der Hals-, Brust- oder Lendenwirbelsäule (je nach dem Ort des Beginns der Symptome) ist Standard, zusätzliche Röntgenuntersuchungen, z.B. vom Brustkorb oder der Wirbelsäule, können notwendig werden.
Eine genetische Untersuchung aus dem Blut ist vor allem sinnvoll bei besonders frühem Beginn der Erkrankung oder wenn andere ähnliche Fälle in der Familie bekannt sind. Mit dem Aufkommen neuer Behandlungsmöglichkeiten für einige häufige Formen der genetisch bedingten ALS gewinnt diese Diagnostik aber auch darüber hinaus an Bedeutung.
In der Regel empfiehlt sich für die Erstdiagnostik ein kurzer stationärer Aufenthalt. Danach werden ambulante Verlaufsuntersuchungen in 3-monatigen Abständen empfohlen.
Weiterführende Infos
Im Download finden Sie weitere Informationen zur Diagnosestellung und zu Behandlungsmöglichkeiten der ALS. Ein Faltblatt informiert zur Physiotherapie bei ALS, ein weiteres kann im Kontakt mit Kostenträgern und Behörden helfen, den zuständigen Mitarbeitenden die Erkrankung und ihre Folgen für den Betroffenen näher zu bringen.
ALS - mit der Krankheit leben lernen
Wenn Sie dies lesen, liegt unter Umständen bei Ihnen oder einem / einer Angehörigen der Verdacht auf ALS oder auf eine Motoneuronerkrankung vor. Möglicherweise haben Sie bereits einen langen Diagnosemarathon hinter sich.
Die Amyotrophe Lateralsklerose (ALS) ist eine voranschreitende Erkrankung, die mit fortlaufenden Veränderungen des Lebensalltages einhergeht. Das betrifft die erkrankte Person besonders, aber auch die Menschen in ihrer unmittelbaren Umgebung.
DGM-Handbuch zur ALS
In unserem Patientenratgeber greifen wir Themen auf, die im Alltag mit der Erkrankung relevant sind. Ziel ist, Ihnen Informationen aus zuverlässigen Quellen gebündelt zur Verfügung zu stellen.
Die ALS-Erkrankung kann sehr unterschiedlich verlaufen. Nicht alles hier Beschriebene trifft bei jedem ein und auch nicht in einer bestimmten Reihenfolge und Geschwindigkeit. Wir empfehlen Ihnen, diesen Informationen als Sammlung zu betrachten und bei Bedarf den jeweils für Sie wichtigen Abschnitt zu Rate zu ziehen.
Sie können das ALS-Handbuch nachfolgend auch als PDF herunterladen oder kostenlos in Papierform in unserem Shop bestellen.
Forschung zur ALS
Weltweit wird sehr intensiv an der Erforschung der Ursachen der ALS und zum Thema Frühdiagnostik gearbeitet. Auch deutsche Forschergruppen, die dem Medizinisch-Wissenschaftlichen Beirat der DGM angehören, sind in diesem Bereich aktiv und haben beste internationale Kontakte. Die Hoffnung, dass in absehbarer Zeit weitere wirksame Behandlungsmethoden zur Verfügung stehen, die entweder direkt an der Ursache oder auch an den Symptomen angreifen, ist berechtigt.
Da neuromuskuläre Erkrankungen und auch ALS zu selten sind, als dass ihre Erforschung wirtschaftlich interessant wäre, spielt die Forschungsförderung der DGM eine bedeutende Rolle: Sie vergibt Forschungsgelder an ausgesuchte Projekte, verleiht Forschungspreise an engagierte Wissenschaftler und arbeitet aktiv in weltweiten neuromuskulären Netzwerken mit.
Infos und Selbsthilfeangebote
Unsere Diagnosegruppe vertritt die Interessen der Betroffenen und Angehörigen.
ALS-Gruppe in der DGMIce Bucket Challenge

Vielen ist die Ice Bucket Challenge, eine Benefizaktion, die 2014 um die Welt ging, noch ein Begriff. Dabei sammelte alleine die DGM Spenden in Höhe von 1,3 Millionen Euro, um die Lebenssituation von Betroffenen zu verbessern und mehr Mittel zur Erforschung der ALS zu generieren.
Unsere Challenge – der Kampf gegen ALS und Hilfe für die Betroffenen – geht weiter.
Wir freuen uns, wenn Sie uns bei unserer Aufgabe unterstützen.