Abschlussbericht zum Forschungsaufenthalt am Queen Square Neuromuscular Centre London, UK
Thema:
Phänotypische Variabilität, Biomarker und klinischer Verlauf der hereditären sensomotorischen Neuropatie -
Evaluation klinischer Scores anhand atypischer CMT-Patienten des Queen Square Neuromuscular Centres (Universitiy College Hospital London) und Etablierung eines international standardisierten Verlaufsprotokolls für das CMT-Register Berlin
Dieses Projekt wurde durch die Förderung der DGM ermöglicht.
Einführung
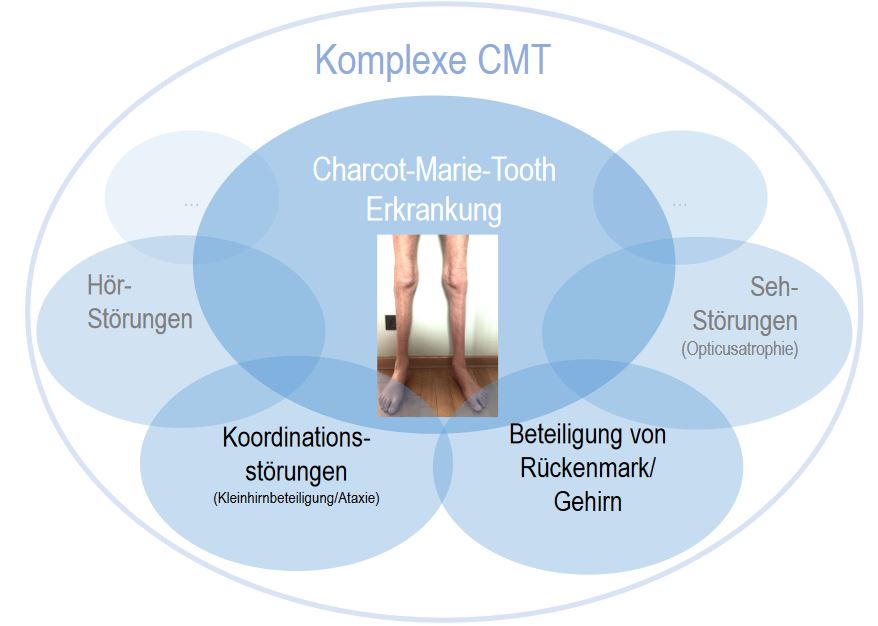
Die international nach den Erstbeschreibern Charcot, Marie, und Tooth (CMT) benannte Erkrankung wird hierzulande auch häufig als hereditäre (also vererbte) motorisch-sensorische (also sowohl die Nerven, die die Muskelbewegung steuern als auch die, die das Fühlen vermitteln) Neuropathie (also Nervenerkrankung), kurz HMSN, bezeichnet. Wie der Name also schon beschreibt, geht es um eine Erkrankung der Nerven, die sowohl die Fähigkeit der Bewegung von Armen und Beinen, als auch das Gefühl, beeinträchtigen kann. Dies beginnt oft schon im Kindesalter und schreitet langsam fort, wobei sich die Muskeln langsam abbauen (Atrophie) und gleichzeitig ein dumpfes, pelziges, oder auch schmerzhaftes Gefühl von Finger- und Zehenspitzen bis hin zum Körperstamm aufsteigen kann. Allerdings stellt es sich heraus, dass in manchen Fällen auch andere Symptome, beispielsweise die Sehfähigkeit (Opticusatrophie) oder die Koordination (Ataxie), vorkommen können. Da das Spektrum der ursächlichen Gene immer größer wird und immer mehr Überschneidungen mit anderen neurologischen Erkrankungen vorkommen, ist es wichtig, die Symptome richtig einordnen zu können und auch den Verlauf der verschiedenen Subtypen zu verstehen.
Die große Diversität an Symptomen, die CMT mit sich bringen kann, birgt nicht nur für die Diagnose, sondern auch für die Behandlung und Therapieentwicklung eine große Herausforderung. International werden verschiedene Skalen und Punktsysteme verwendet, um den Verlauf von Erkrankungen objektiv nachzuverfolgen. Das Queen Square Neuromuscular Centre in London (QSNMC) ist eines der größten und renommiertesten Zentren für die Behandlung von CMT europa- und weltweit. Hier wurden einige der heutzutage wichtigsten Genentdeckungen gemacht, aber auch Studien zum natürlichen Krankheitsverlauf und zu den Behandlungsmethoden entwickelt. Darüber hinaus besteht ein wichtiges Netzwerk weltweiter Spezialisten. Es ist sehr wichtig, dass die Spezialisten für CMT weltweit möglichst einheitliche Standards verwenden, um Patientinnen und Patienten mit CMT zu beurteilen, damit die ermittelten Daten miteinander verglichen werden können. Denn nur so kommen bei dieser seltenen Krankheit am Schluss genug Patienten zusammen, um sinnvolle Studien zu Therapien durchzuführen und den Effekt möglicher Behandlungen auch wirklich zu erfassen.
Worum ging es in diesem Projekt?
Ziel des 6-monatigen Projekts am QSNMC war es auf der einen Seite, international einheitliche Standards zu erarbeiten, und auf der anderen Seite, CMT-Patienten mit komplexen Symptomen zu charakterisieren und wichtige Aspekte zu identifizieren, um diese in einheitliche Verlaufsuntersuchungen einzubeziehen.
Was sind erste Ergebnisse?
Zu den wichtigsten Verlaufsuntersuchungen gehören der CMT Neuropathy Score (CMTNS) sowie verschiedene Patientenfragebögen, sowie laborchemische Marker wie zum Beispiel Neurofilament-Leichtketten. Hieraus wurde ein Protokoll für die CMT-Sprechstunde an der Charité Berlin etabliert und soll die zukünftige Vernetzung und Teilung von Daten mit anderen CMT-Registern ermöglichen.
In der Analyse der Patienten mit komplexer CMT zeigten besonders viele Patienten Zeichen des ersten Motorneurons. Das bedeutet, dass in manchen Unterformen der CMT neben den peripheren Nerven in Armen und Beinen auch am Rückenmark oder im Gehirn Schäden auftreten können. Darüber hinaus hatten mehrere Patienten Zeichen einer Kleinhirnbeteiligung, die sich auf die Koordination und die Augenbewegung auswirkt, andere hatten Hör- oder Sehstörungen, oder Lernschwäche. Dazu trugen speziell Mutationen mitochondrialer Gene bei, also Veränderungen in den „Energiefabriken“ der Zellen. Bei Patienten mit komplexer CMT konnte außerdem in einem wesentlich größeren Anteil kein ursächliches Gen gefunden werden als bei Patienten mit klassischer CMT – was dafürspricht, dass hier komplexe genetische Ursachen oder neue, noch nicht entdeckte Gene, zugrunde liegen.
Wie geht es weiter?
Aufbauend auf den bisherigen Ergebnissen sollen neue krankheitsverursachende Gene identifiziert und neue Aspekte bekannter Subtypen der CMT beschrieben werden. Hierfür werden Patientinnen und Patienten mit CMT in unserer Registerstudie an der Charité gesammelt, anhand des entwickelten Protokolls untersucht, und die Vernetzung mit anderen Registern geplant. Außerdem entwickeln wir aufbauend auf den bereits bekannten Methoden neue Technologien zur genaueren und einfacheren Verlaufsuntersuchung.
Was bedeutet das für Betroffene?
Ein Patient mit der Verdachtsdiagnose CMT sollte eine Anbindung an ein Zentrum mit CMT-Spezialisierung finden, in dem internationale Skalen verwendet und regelmäßige Verlaufsuntersuchungen angeboten werden. Denn nur durch das Zusammenführen der Befunde vieler Patienten können krankheitsassoziierte Merkmale identifiziert und deren Ursachen gefunden werden. So kann jeder einzelne dazu beitragen, dass die vielversprechenden neuen Therapieansätze aus der Forschung bald Realität werden.
Dr. med. Helena F. Pernice
CMT-Sprechstunde der Charité Berlin
Amyloidosis Center Charité Berlin (ACCB)
Klinik für Neurologie und Experimentelle Neurologie
Charité – Universitätsmedizin Berlin
