Spinale Muskelatrophie (SMA)
Erklärvideo Spinale Muskelatrophie (SMA)
 Öffnet Video in Overlay
Öffnet Video in Overlay
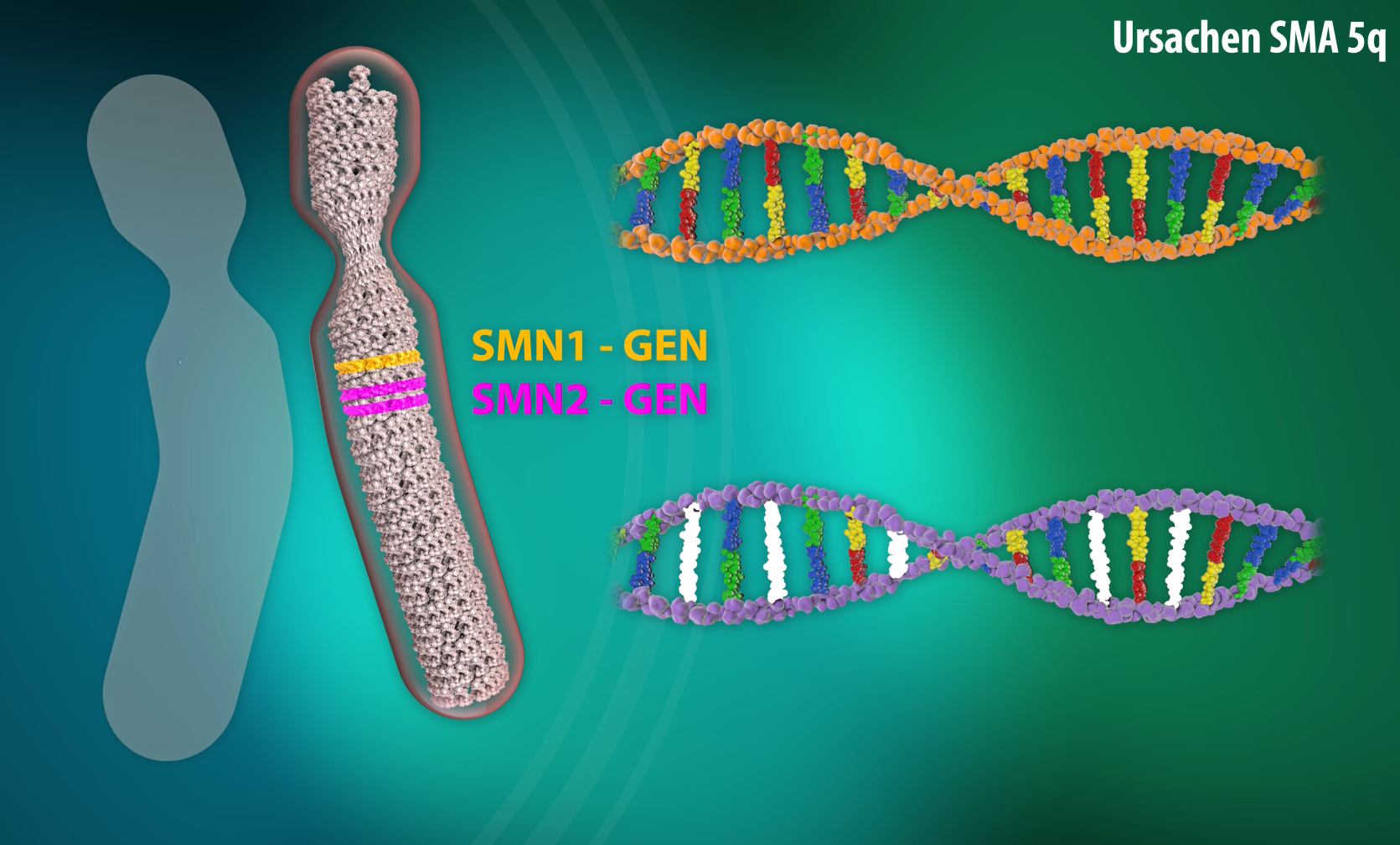
Die Spinale Muskelatrophie 5q (SMA) ist eine Motoneuronerkrankung, d.h. eine Erkrankung bestimmter Nervenzellen im Rückenmark. Diese Nervenzellen leiten Impulse an die Muskulatur weiter, die für die willkürlichen Bewegungen wie Krabbeln, Laufen und Kopfkontrolle zuständig sind. „5q“ bedeutet, dass die genetische Veränderung in einer bestimmten Region auf dem Chromosom 5 gefunden werden konnte.
Die Spinale Muskelatrophie ist eine relativ häufige „Seltene Erkrankung“: Ungefähr eines von 6 -10.000 Neugeborenen ist betroffen und ungefähr eine von 45 Personen ist Überträger der Erkrankung.
Symptome der SMA
SMA beeinträchtigt alle Muskeln des Körpers, wobei die sogenannten proximalen Muskeln (die dem Rumpf am nächsten sind, z.B. Schulter-, Hüft- und Rückenmuskulatur) am schwersten betroffen sind. Die Schwäche in den Beinen ist im Allgemeinen größer als in den Armen. Es kann auch die Kau- und Schluckmuskulatur betroffen sein. Die Beteiligung der Atemmuskulatur, die für die Aufnahme von Sauerstoff, die Abgabe von CO2 und das Abhusten zuständig ist, kann zu einer erhöhten Anfälligkeit für Lungenentzündungen und zu anderen Problemen mit der Lunge führen. Sinneswahrnehmungen, das heißt Sehen, Hören, Riechen, Schmecken und die Hautsensibilität sind nicht betroffen. Die intellektuellen Fähigkeiten sind ebenfalls nicht betroffen. Es wird im Gegenteil oft beobachtet, dass Patienten mit SMA geistig wach und kontaktfreudig sind.
Patienten mit SMA verlieren typischerweise im Verlauf der Erkrankung an Muskelkraft. Das kann im Rahmen eines Wachstumsschubes bei Kindern sehr rasch passieren oder auch ganz allmählich stattfinden. Es kann sein, dass Patienten mit SMA oft über einen längeren Zeitraum relativ stabil in ihren motorischen Funktionen sind. Die Tendenz zum Verlust von Funktionen ist jedoch immer vorhanden und bleibt auch im Erwachsenenalter bestehen.
Therapieoptionen
Es existieren in Deutschland unterdessen drei zugelassene Medikamente für die Behandlung der SMA. Alle drei Therapieoptionen unterscheiden sich im Wirkmechanismus, Zulassungslabel sowie Verabreichungsform. Daher ist eine fachliche Beratung durch erfahrene Behandelnde dringend erforderlich.
Behandlungs- und Beratungszentren für SMA
Auf www.dgm-behandlungszentren.org finden Sie eine interaktive Landkarte mit den Behandlungs- und Beratungszentren für SMA-Betroffene aller Altersgruppen.
DGM-BehandlungszentrenWeiterführende Infos
Im Download finden Sie Informationen zur Symptomatik und zu den verschiedenen Formen der SMA, zur Diagnosestellung sowie zur medikamentösen Therapie und weiteren Behandlungsmöglichkeiten. Auch Hinweise zu den Besonderheiten der Physiotherapie bei SMA können Sie hier herunterladen.
Handlungsempfehlungen zur Behandlung der spinalen Muskelatrophie mit Zolgensma®
Das Konsensuspapier wurde auf Einladung DGM unter Beteiligung der deutschen neuromuskulären Behandlungszentren, der deutschen Sektion der Gesellschaft für Neuropädiatrie (GNP) und unter Mitwirkung des Medizinisch-Wissenschaftlichen Beirates der DGM 2020 erarbeitet. Ziel ist es, die notwendigen Voraussetzungen für eine qualitätsgesicherte Anwendung der neuen Gentherapie zu definieren und die Grundlage für die Umsetzung in der klinischen Praxis zu schaffen.
Leitfaden zu den Behandlungsstandards
Newbornscreening auf SMA
ab 1. Oktober 2021
Bei der spinalen Muskelatrophie (SMA) fehlt den Kindern ein Faktor („survival motor neuron“-Faktor, SMN), der notwendig für das Überleben von speziellen Nervenzellen ist. Betroffen sind die sogenannten Motoneurone, die für die Steuerung der Muskulatur verantwortlich sind. Durch diesen Mangel gehen diese Nervenzellen zugrunde. In der Folge kommt es zu einer meist schweren Muskelschwäche. Wird der Defekt frühzeitig entdeckt, stehen inzwischen mehrere verschiedene Therapiemöglichkeiten zur Verfügung, um dem Körper ausreichend SMN zur Verfügung zu stellen. Die Nervenzellen können dann überleben und die Kinder können sich im besten Falle sogar normal entwickeln. Entscheidend ist die Entdeckung der Erkrankung noch bevor Symptome auftreten.
Auf www.dgm-behandlungszentren.org finden Sie Ihr nächstgelegenes Beratungszentrum zum Neugeborenen-Screening auf SMA sowie auch die Therapiezentren für die drei derzeit in Deutschland zugelassenen Medikamente Spinraza®, Evrysdi® und Zolgensma®.
Wenige Tage nach der Geburt wird meist noch in der Geburtsklinik dem Kind aus der Ferse etwas Blut abgenommen. In dieser winzigen Blutprobe wird dann nach einer ganzen Reihe von Erkrankungen gesucht. Sollten sich Hinweise auf eine SMA ergeben, wird die Familie entweder von der Geburtsklinik, dem Labor oder einem Neuromuskulären Zentrum (NMZ) angerufen, um sich innerhalb weniger Tage im Behandlungszentrum vorzustellen.
Das Kind wird untersucht und es wird nochmals eine Blutprobe für eine zweite Laboruntersuchung entnommen, um ganz sicher zu gehen, dass die Diagnose stimmt. Aus dieser Blutprobe werden gleichzeitig noch weitere Informationen gewonnen, die wichtig für die Entscheidung sind, wie rasch die Behandlung beginnen muss. Die Experten im NMZ besprechen mit den Eltern nach Erhalt der Befunde im Detail, welche Therapien in Frage kommen.
Die Daten, die bisher in Studien gewonnen wurden, zeigen, dass sich die früh behandelten Kinder dramatisch besser entwickeln, verglichen mit dem Verlauf der Erkrankung, wie wir ihn früher sehen mussten.
Die Diagnostik und die Therapie werden in Deutschland erfreulicherweise von den Krankenkassen übernommen. Die Auswahl des Medikaments erfolgt entsprechend den Zulassungskriterien der verschiedenen Substanzen.
Infos und Selbsthilfeangebote
Unsere Diagnosegruppe vertritt die Interessen der Betroffenen und Angehörigen.
SMA-Gruppe in der DGM