Muskeldystrophien Duchenne und Becker
Erklärvideo Muskeldystrophien Duchenne und Becker
 Öffnet Video in Overlay
Öffnet Video in Overlay
Erkrankungsursache
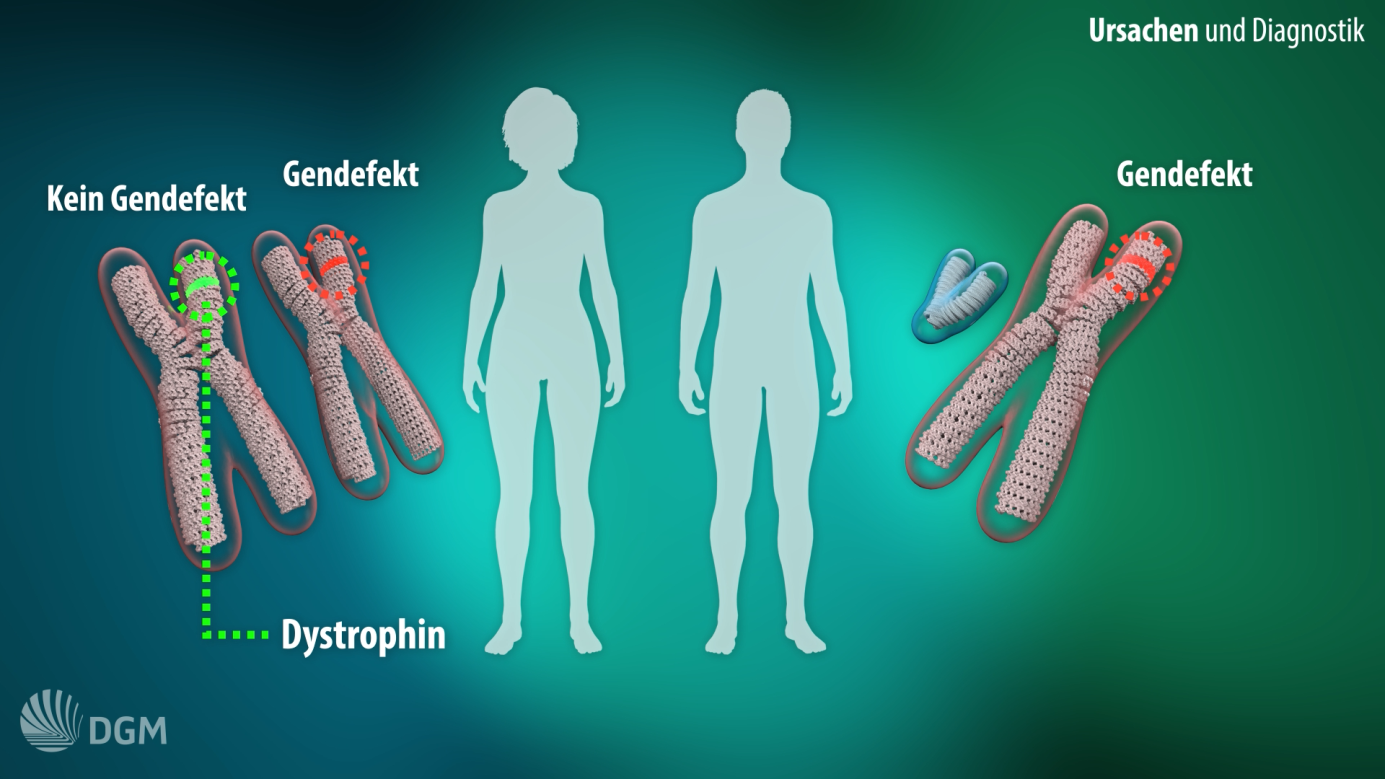
Die häufigste Ursache für eine Muskeldystrophie (MD) im Kindes- und Jugendalter ist ein erblich bedingtes Fehlen oder eine Funktionsänderung des Muskel-Eiweißes Dystrophin. Betroffen sind fast ausschließlich Jungen.
Muskeldystrophie Duchenne
Vollständiges Fehlen von Dystrophin führt zur Muskeldystrophie Duchenne (DMD), die mit einer Häufigkeit von einem von 3.500 - 5.000 neugeborenen Jungen auftritt. In Deutschland leben 1.500 bis 2.000 Betroffene.
Muskeldystrophie Becker
Eine Verkürzung und Funktionseinschränkung des Dystrophins führt zur milderen und langsamer fortschreitenden Becker-Muskeldystrophie (BMD). Sie ist im Kindesalter die seltenere Dystrophin-Erkrankung, dennoch bei älteren Jugendlichen und Erwachsenen die mit großem Abstand häufigste Muskeldystrophie.
Weiterführende Infos
Im Download finden Sie Informationen zur Ursache und Symptomatik, zur Diagnosestellung und zu therapeutischen Möglichkeiten bei den beiden Dystrophin-Erkrankungen. Auch Hinweise zu den Besonderheiten der Physiotherapie bei BMD / DMD sowie einen ausführlichen Familienratgeber können Sie hier herunterladen.
Einfach erklärt: Arzneimittelforschung bei Muskeldystrophie Duchenne (DMD)
 Öffnet Video in Overlay
Öffnet Video in Overlay
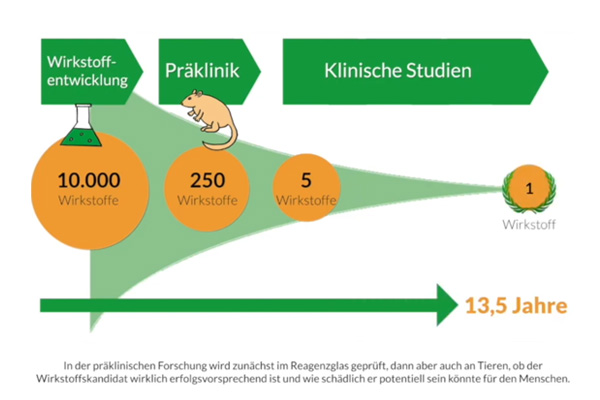
Wie ist der Stand der Medikamentenentwicklung bei der Muskeldystrophie Duchenne? Wie funktioniert die Entwicklung neuer Medikamente und warum dauert sie mehrere Jahre?
Eine Ärztin eines von der Deutschen Gesellschaft für Muskelkranke e.V. (DGM) zertifizierten Neuromuskulären Zentrum gibt verständliche Antworten auf die spannenden Fragen betroffener Familien.
Infos und Selbsthilfeangebote
Unsere Diagnosegruppe vertritt die Interessen der Betroffenen und Angehörigen.
DMD/BMD-Gruppe (Duchenne/Becker-Kiener) in der DGM