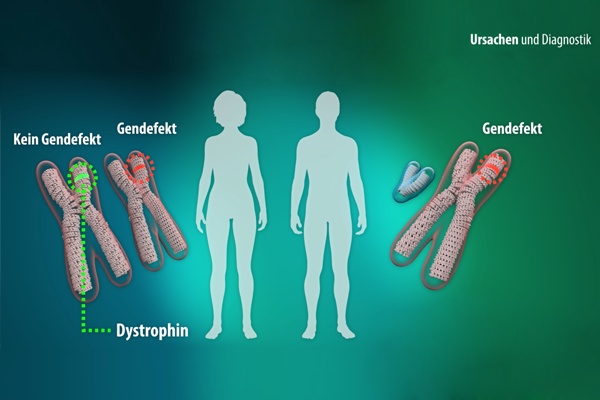
Mehr als 100.000 Menschen in Deutschland sind von einer Muskelerkrankung betroffen. Viele haben schon von "Muskelschwund" gehört, aber was ist das eigentlich? Welche verschiedenen Muskelkrankheiten gibt es?
Veschaffen Sie sich einen Überblick über Ursache & Symptome, Diagnose & Therapie sowie zum aktuellen Stand der Forschung.



